Метод Гюккеля
Метод Гю́ккеля або молекулярна орбітальна теорія Гюккеля (англ. Hückel molecular orbital theory) — різновид методу МО ЛКАО для визначення енергій π-орбіталей у делокалізованих π-системах (напр. етилен, бензен, бутадієн).[1][2] Метод був уперше запропонований Еріхом Гюккелем у 1930 р. як теоретична основа для правила Гюккеля, а пізніше був розширений для гетероциклічних сполук, таких як піридин, пірол або фуран.[3] Подальше розширення методу Гюккеля, відоме як розширений метод Гюккеля (англ. extended Hückel method, EHM), також бере до уваги σ-електрони. Цей метод був розроблений Роалдом Гоффманом як теоретичний апарат для правил Вудварда–Гоффмана.[4] Щоб запобігти плутанині, класичний метод Гюккеля тепер також називають простим методом Гюккеля (англ. simple Hückel method, SHM).
Незважаючи на свою відносну математичну простоту й обмеженість молекул, до яких він може бути застосований, метод Гюккеля у своєму первісному вигляді є доволі потужним інструментом для аналізу властивостей кон'югованих π-систем. Опис методу Гюккеля можна зустріти в багатьох вступних підручниках з квантової хімії та фізичної органічної хімії; в органічній хімії ця теорія й досі активно застосовуєтьтся для передбачення результату реакцій, де беруть участь кон'юговані π-зв'язки (наприклад, перициклічних реакцій).
Математична основа методу
Метод Гюккеля використовує роздільність σ-π-електронів: увага приділяється лише π-МО, оскільки саме вони визначають значну частину хімічних і спектральних властивостей молекул. Конструювання π-МО відбувається тільки з 
Для кон'югованих лінійних полієнів (наприклад, 1,3,5-гексатрієн), енергетичні рівні π-МО задані рівнянням:

де


із числом атомів Карбону 
Величина 


Для повністю делокалізованих циклічних полієнів застосовується формула:



Лінійні полієни
Знаючи енергії орбіталей, можна сконструювати діаграму МО; ті орбіталі, для яких 



Нижче наведені діаграми МО для етилену, бутадієну й гексатрієну (червоним позначені вузлові площини):[6]

Для π-систем з непарною кількістю атомів Карбону діє такий же принцип, але кількість МО в такому випадку теж буде непарною, а одна орбіталь буде незв'язуючою:[6]

Циклічні полієни: Цикл Фроста
Вид діаграм МО для спряжених циклічних полієнів лежить в основі так званого циклу Фроста-Музуліна (зазвичай використовують назву "цикл Фроста"). Розрахунок енергій МО, як це описано вище, приводить до цікавого спостереження, що циклічні полієни з 

- Приклади ароматичних сполук
 Циклопропеніл-катіон
Циклопропеніл-катіон Циклопентадієніл-аніон
Циклопентадієніл-аніон Циклогептатрієніл-катіон (тропілій-катіон)
Циклогептатрієніл-катіон (тропілій-катіон)
_with_MO_diagram.png)
_with_MO_diagram.png)
_with_MO_diagram.png)
- Приклади антиароматичних сполук
 Циклобутадієн
Циклобутадієн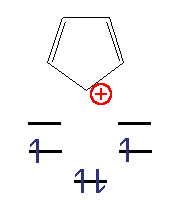 Циклопентадієніл-катіон
Циклопентадієніл-катіон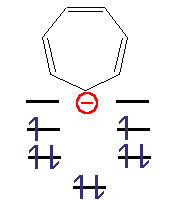 Циклогептатрієніл-аніон
Циклогептатрієніл-аніон

_with_MO_diagram.png)
_with_MO_diagram.png)
{kind=link}
{kind=link}