Variants du SARS-CoV-2
Le SARS-CoV-2 (coronavirus 2 du syndrome respiratoire aigu sévère) est le virus qui cause la maladie à coronavirus 2019 (Covid-19). La séquence WIV04 / 2019 serait la séquence d'origine infectant les humains[1]. Comme tout virus, le SARS-CoV-2 a évolué et a créé des variants. Certains sont considérés comme d'une importance particulière. D614G est probablement la mutation la plus importante et l'une des plus répandues du SARS-CoV-2. Cette mutation empêche un mécanisme d'infection des globules blancs connu sous le nom de facilitation de l'infection par des anticorps.

Cet article doit être actualisé ().
Des passages de cet article ne sont plus d’actualité ou annoncent des événements désormais passés. Améliorez-le ou discutez-en. Vous pouvez également préciser les sections à actualiser en utilisant {{section à actualiser}}.
Wikipédia ne donne pas de conseils médicaux ou sanitaires. Cet article est susceptible de contenir des informations obsolètes ou inexactes. Seul un professionnel de la santé est apte à vous fournir un avis médical, et seules les autorités sanitaires de votre pays sont compétentes pour donner des consignes de santé publique relatives à la pandémie de Covid-19.
Les coronavirus sont censés être stables dans la mesure où ils produisent une enzyme correctrice d’erreurs appelée « exoribonucléase ». D’après l’INSERM, le SARS-CoV-2 muterait ainsi deux fois moins rapidement que les virus grippaux[2]. Dans les faits, le SARS-CoV-2 se révèle assez mutagène[3],[4].

Fin 2020 et début janvier 2021, trois variants problématiques sont identifiés : le variant alpha en Angleterre, le variant bêta en Afrique du Sud, et le variant gamma au Brésil. En 2021, au moins sept autres variants ont été recensés par l’OMS comme d’intérêt. Les variants les plus contagieux (de 50 à 75 % par rapport à la souche initiale) ont été successivement le variant alpha durant l', puis le variant delta identifié en Inde à partir du .
Fin 2022, de nouveaux variants dérivant de la souche Omicron circulent dans le monde. : BQ.1.1 en Europe ou XBB en Asie sont "surveillés du fait de leurs mutations sur la protéine Spike, leur conférant un potentiel pouvoir d’échappement immunitaire"[5].
Ces variants ne remettent pas en cause l’efficacité des vaccins contre la Covid-19. Seules des combinaisons de mutations bien spécifiques sur la protéine S sont susceptibles en théorie de compromettre l'efficacité des vaccins. Une combinaison déjà identifiée pourrait être le trio de mutations[6],[7] : F140-, Y248* et E484K.
Nomenclature
Aucune nomenclature pour les lignées évolutives du SARS-CoV-2 n'est universellement acceptée[8], cependant en , l’Organisation mondiale de la santé travaille sur une « nomenclature standard pour les variants du SARS-CoV-2 qui ne fait pas référence à une localisation géographique »[9]. Dans une lettre d'information datée du , l'OMS annonce nommer les variants par une lettre grecque[10]. L'OMS précise que « ces noms ne remplacent pas les noms scientifiques [attribués par d'autres], qui véhiculent des informations scientifiques importantes ».
Bien qu'il existe plusieurs milliers de variants du SARS-CoV-2[11], les sous-types du virus peuvent être placés dans des groupes beaucoup plus larges tels que les lignées ou les clades. Plusieurs nomenclatures différentes pour ces sous-types ont été proposées.
Catégorisation des variants
- variant préoccupant[12], VOC (variant of concern)
- variant d'intérêt[12], ou variant à suivre[12], VOI (variant of interest) également dénommé VUI (variant under investigation)[13]
- variant en cours d’évaluation, ou variant sous surveillance[12], VUM (variant under monitoring)
Différentes nomenclatures
Entités réalisant un nommage
Nextstrain
Nextstrain nomme les clades majeurs par le numéro de l'année et une lettre (les lettres sont successives, depuis « A », chaque nouvelle année)[14], par exemple : 19A, 19B, 20A.Pour un clade émergent, la notation comporte alors le nom du clade parent, et la mutation de nucléotide qui le définit[14] (attention, la numérotation est par nucléotide, et non par codon), par exemple, 19A/28688C signifie : pour le clade 19A, en position de nucléotide 28688, la base nucléique est maintenant la cytosine (C).Une fois que le clade émergent a atteint les critères nécessaires, il est renommé comme clade majeur[14], par exemple : 19A/28688C devient 20D.
Les règles de nommage ont été révisées début [15] pour éviter les nommages géographiques tel 20A.EU1 (variant émergent en Europe). Une notation mixte 20H/501Y.V2 ou 20I/501Y.V1 est alors utilisée avec un numéro d'ordre : V1, V2, etc.
Lors de cette révision des règles, les noms des clades ont été conservés (rétrocompatibilité), sauf pour trois noms :
- 20A.EU1 devient « 20E (EU1) » – 20A.EU1 ayant été élevé au rang de clade (il devrait alors se nommer « 20E » uniquement) ; les parenthèses « (EU1) » établissent le lien entre l'ancien et le nouveau nom[15] ;
- 20B/501Y.V1 devient 20I/501Y.V1 – nommage plus complexe : 20B/501Y.V1 ayant été élevé au rang de clade (nommé « 20I » uniquement), mais ayant été précédemment défini avec le nom « 501Y.V1 » seul[16], cette même terminaison a été conservée, sans provoquer de confusion[15],[17] ;
- 20C/501Y.V2 devient 20H/501Y.V2 – idem.
Le nom d'un clade émergeant s'effectue avec le nom du clade parent et, selon le numéro de nucléotide, la mutation qui le définit[14], par exemple : 20G/1927C signifie un changement de nucléotide (ici en cytosine – C) à la position 1927, dans le clade 20G.Cependant, afin de pouvoir nommer des mutations au niveau des codons, une règle a été définie pour attribuer des noms par mutation dans chaque cadre de lecture ouvert (ORF) codant chaque protéine[15], par exemple 20B/S.484K signifie une mutation du codon (ici codage de la lysine – K) à la position 484 dans la protéine S (notation « S. ») du virus. Les numéros de codons sont relatifs à chaque ORF.

Les derniers clades définis (en ) sont 23G, 23H, 23I (Omicron : XBB.1.5.70 ; HK.3 ; BA.2.86).
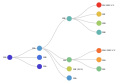 Clades Nextstrain (2019-2020).
Clades Nextstrain (2019-2020).
Début , les clades alors en usage étaient :
20A, 20B, 20C, 20D, 20E, 20F, 20G, 20H/501Y.V2 et 20I/501Y.V1.

Depuis les noms attribués par l'OMS sont alors indiqués :
 Clades Nextstrain, .
Clades Nextstrain, . Clades Nextstrain, .
Clades Nextstrain, . Clades Nextstrain, .
Clades Nextstrain, . Clades Nextstrain, .
Clades Nextstrain, .




Depuis , les lignées Pango sont également indiquées :
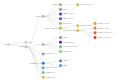 Clades Nextstrain, (+ 22D).
Clades Nextstrain, (+ 22D).- Clades Nextstrain, (+ 22E, 22F).
 Clades Nextstrain, (+ 23A).
Clades Nextstrain, (+ 23A). Clades Nextstrain, (+ 23B).
Clades Nextstrain, (+ 23B). Clades Nextstrain, (+ 23C, 23D, 23E, 23F).
Clades Nextstrain, (+ 23C, 23D, 23E, 23F). Clades Nextstrain, (+ 23G, 23H, 23I).
Clades Nextstrain, (+ 23G, 23H, 23I).




GISAID
En mars 2021, GISAID avait défini huit clades[18] pour les variants du SARS-CoV-2. Les clades sont nommés d'après leur mutation essentielle. Depuis les deux clades initiaux, « S » et « L » (ces noms proviennent d'une étude préalable à celle de GISAID[18],[19]), les autres clades sont :
- S
- L
- V (pour NS3-G251V)
- G (pour S-D614G)
- GR (pour N-G204R)
- GRY (pour S-N501Y)
- GH (pour NS3-Q57H)
- GV (pour S-A222V)
- GR (pour N-G204R)
Ces clades GISAID peuvent alors être affinés par la définition de lignées, comme celles de Lignées Pango.
Lignées Pango
Le système de nomenclature « Lignées Pango »[20] est un système standardisé et dynamique, désignant les variants alarmants du SARS-CoV-2 en fonction de la phylogénie[21],[22].
- A [2019-12-30] (second haplotype découvert – après « B » – ; la racine de la pandémie se situe dans cette lignée A).
- A.23
- A.23.1 [2020-10-21]
- B [2019-12-24] (premier haplotype découvert).
- B.1 [2020-01-24]
- B.1.1 [2020-02-16]
- B.1.1.1
- C.1, alias de B.1.1.1.1
- C.1.2, [2021-05-11], alias de B.1.1.1.1.2
- C.36, alias de B.1.1.1.36
- C.36.3, alias de B.1.1.1.36.3
- C.36.3.1 [2021-03-27], alias de B.1.1.1.36.3.1
- C.36.3, alias de B.1.1.1.36.3
- C.37 [2020-11-08], alias de B.1.1.1.37 (Lambda)
- B.1.1.7 [2020-02-07] (Alpha)
- Q.* [2020-07-11] à [2021-03-02], alias de B.1.1.7.*
- B.1.1.28
- P.1 [2020-04-07], alias de B.1.1.28.1 (Gamma)
- P.1.1 [2021-01-07], alias de B.1.1.28.1.1
- P.1.2 [2021-01-14], alias de B.1.1.28.1.2
- P.1.4 [2021-03-19], alias de B.1.1.28.1.4
- P.1.6 [2021-03-26], alias de B.1.1.28.1.6
- P.1.7 [2021-03-10], alias de B.1.1.28.1.7
- P.2 [2020-04-13], alias de B.1.1.28.2 (Zêta)
- P.3 [2021-01-08], alias de B.1.1.28.3 (Thêta)
- P.1 [2020-04-07], alias de B.1.1.28.1 (Gamma)
- B.1.1.316
- R.1 [2020-01-14], alias de B.1.1.316.1
- R.2 [2020-12-22], alias de B.1.1.316.2
- B.1.1.318 [2021-01-07]
- B.1.1.370
- AT.1 [2021-01-18], alias de B.1.1.370.1
- B.1.1.482
- AV.1 [2021-02-18], alias de B.1.1.482.1
- B.1.1.519 [2020-07-22]
- B.1.1.529 [2021-11-08] (Omicron)
- BA.1 [2021-01-12], alias de B.1.1.529.1 (Omicron)
- XD [2022-01-03], recombinant de Delta – lignée AY.4 (B.1.617.2.4) et BA.1 — voir également AY.4 (Delta)
- XE [2022-01-19], recombinant de BA.1 et BA.2 — voir également BA.2 (Omicron)
- BA.2 [2021-11-17], alias de B.1.1.529.2 (Omicron)
- XE [2022-01-19], recombinant de BA.1 et BA.2 — voir également BA.1 (Omicron)
- BA.3 [2021-11-25], alias de B.1.1.529.3 (Omicron)
- BA.4 [2022-01-10], alias de B.1.1.529.4 (Omicron)
- BA.5 [2022-01-06], alias de B.1.1.529.5 (Omicron)
- BA.1 [2021-01-12], alias de B.1.1.529.1 (Omicron)
- B.1.1.1
- B.1.177 [2020-02-14]
- B.1.214
- B.1.214.2 [2020-11-22]
- B.1.351 [2020-05-09] (Bêta)
- B.1.351.1 [2021-01-19]
- B.1.351.2 [2020-12-06]
- B.1.351.3 [2021-01-12]
- B.1.427 [2020-04-11]
- B.1.429 [2020-03-11]
- B.1.466
- B.1.466.2 [2020-08-06]
- B.1.525 [2020-12-11] (Êta)
- B.1.526 [2020-04-21] (Iota)
- B.1.617
- B.1.617.1 [2020-12-01] (Kappa)
- B.1.617.2 [2020-09-07] (Delta)
- AY.1 [2021-04-05], alias de B.1.617.2.1
- AY.2 [2021-03-12], alias de B.1.617.2.2
- AY.3 [2021-04-23], alias de B.1.617.2.3
- AY.3.1 [2020-11-20], alias de B.1.617.2.3.1
- AY.4 [2020-05-11], alias de B.1.617.2.4
- XD [2022-01-03], recombinant de Delta – lignée AY.4 (B.1.617.2.4) et BA.1 — voir également BA.1 (Omicron)
- AY.5 ... AY.12 [2020-07-13](AY.10) ... [2020-11-15](AY.5), alias de B.1.617.2.5 ... B.1.617.2.12
- AY.13 ... AY.25 [2020-07-21](AY.25) ... [2021-05-30](AY.23.1), alias de B.1.617.2.13 ... B.1.617.2.25
- B.1.619 [2020-03-10]
- B.1.619.1 [2021-02-06]
- B.1.620 [2021-02-06]
- B.1.621 [2021-01-11] (Mu)
- B.1.621.1 [2021-03-18]
- B.1.1 [2020-02-16]
- B.1 [2020-01-24]
- A.23
Public Health England
Lors de la détection d'un variant dans le Kent au sud-est de Londres, l'agence exécutive gouvernementale britannique Public Health England (PHE) a défini une nomenclature.
En septembre 2020, PHE a détecté un variant qui s'est révélé digne d'intérêt en , et l'a appelé, le , « first Variant Under Investigation in December 2020 »[24] (VUI-202012/01, pour Variant Under Investigation, year 2020, month 12, variant 01)[25],[26].Ce variant a été rebaptisé « variant préoccupant » (Variant of Concern, VOC) le , soit alors « VOC-202012/01 »[27].
Mi-, PHE a modifié ses règles de nommage en : [VOC/VUI] – [deux derniers chiffres de l'année] – [trois caractères du mois] – [numéro de variant dans le mois sur deux digits][28].Depuis lors, « VOC-202012/01 » est connu comme « VOC-20DEC-01 ».
Public Health England a également été chargé du séquencement du variant dit « sud-africain », en . Ce variant a alors reçu le nom de « VOC-202012/02 »[29].Il est depuis nommé « VOC-20DEC-02 »[28] (synonymes : 501Y.V2, B.1.351)[30].
Pour les variants dits « brésiliens », Public Health England a effectué le séquençage d'un variant en , depuis nommé « VUI-21JAN-01 »[28] (synonymes : Zêta, ou P.2 – descendant de B.1.1.28)[30], et d'un variant détecté au Japon, sur des voyageurs en provenance du Brésil, depuis nommé « VOC-21JAN-02 »[28] (synonymes : Gamma, ou P.1 – descendant de B.1.1.28)[30].
Noms et équivalences
Depuis le , l'OMS a défini la règle de nommage des variants[10], et catégorisé ces variants (VOC/VOI/VUM)[31]. Dans les tableaux suivants, c'est cette qualification qui est retenue.
Cependant, le nommage des variants, et leur catégorisation évolue dans le temps. Ainsi, l'état est ici indiqué pour la dernière publication de l'OMS connue : au .
- [31] :
- • définition de la règle de nommage[10] ;
- • publication des VOC « Alpha », « Bêta », « Gamma » et « Delta » ;
- • publication des VOI « Epsilon », « Zêta », « Êta », « Thêta », « Iota », « Kappa » ;
- [32] :
- • publication du VOI « Lambda » ;
- [33] :
- • classification de nouvelles lignées, pour les variants Bêta et Gamma :
- outre B.1.351, Bêta est maintenant défini comme B.1.351 + B.1.351.2 + B.1.351.3
- outre P.1, Gamma est maintenant défini comme P.1 + P.1.1 + P.1.2
- • surveillances additionnelles introduites pour les variants Alpha et Gamma :
- pour Alpha, surveillance des mutations S:484K et S:452R ;
- pour Gamma, surveillance de la mutation S:417N ;
- [34] :
- • surveillances additionnelles introduites pour les variants Bêta et Delta :
- pour Bêta, surveillance de la mutation S:L18F ;
- pour Delta, surveillance de la mutation S:417N ;
- • les variants Epsilon, Zêta, Thêta sont requalifiés de VOI à VUM ;
- • les variants VUM sont publiés :
- B.1.427/B.1.429 (ex-
Epsilon) - P.2 (ex-
Zêta) - P.3 (ex-
Thêta) - R.1 & R.2
- B.1.466.2
- B.1.621
- AV.1
- B.1.1.318
- B.1.1.519
- AT.1
- C.36.3 + C.36.3.1
- B.1.214.2
- B.1.427/B.1.429 (ex-
- [35] :
- • classification d'une nouvelle lignée intégrée au variant Delta :
- outre B.1.617.2 + AY.1 + AY.2, Delta est maintenant défini comme B.1.617.2 + AY.1 + AY.2 + AY.3
- • les variants B.1.1.523, B.1.619 et B.1.620 sont ajoutés à la liste des VUM.
- [36] :
- • dans la liste des VUM, retrait de AV.1 & AT.1, qui ne sont maintenant plus suivis ;
- [37] :
- • classification de nouvelles lignées, pour des noms de variants existants :
- outre P.1 + P.1.1 + P.1.2, Gamma est maintenant défini comme P.1 + P.1.1 + P.1.2 + P.1.4 + P.1.6 + P.1.7
- outre B.1.617.2 + AY.1 + AY.2 + AY.2, Delta est maintenant défini comme B.1.617.2 + AY.1 + AY.2 + AY.3 + AY.3.1
- • dans la liste des VUM :
- retrait de P.2 (ex-
Zêta), P.3 (ex-Thêta) et R.2, qui ne sont maintenant plus suivis ; - la surveillance de B.1.619 est étendue à celle de B.1.619 + B.1.619.1 ;
- la surveillance de B.1.621 est étendue à celle de B.1.621 + B.1.621.1 ;
- retrait de P.2 (ex-
- [38] :
- • classification de nouvelles lignées, pour des noms de variants existants :
- outre B.1.1.7, Alpha est maintenant défini comme B.1.1.7 + Q.* (c.-à-d. B.1.1.7.* — « * » signifiant « n'importe quel suffixe » ) ;
- outre B.1.351, Bêta est maintenant défini comme B.1.351 + B.1.351.2 + B.1.351.3 ;
- Gamma, auparavant défini comme P.1 + P.1.1 + P.1.2 + P.1.4 + P.1.6 + P.1.7, est maintenant réduit à P.1 + P.1.4 + P.1.6 + P.1.7 ;
- Delta, auparavant défini comme B.1.617.2 + AY.1 + AY.2 + AY.3 + AY.3.1, est maintenant défini comme B.1.617.2 + AY.* (c.-à-d. B.1.617.2.*) — « * » signifiant « n'importe quel suffixe » ) ;
- [39] :
- • classification de nouvelles lignées, pour des noms de variants existants :
- Gamma, auparavant défini comme P.1 + P.1.4 + P.1.6 + P.1.7, est maintenant réduit à P.1 ;
- • publication du VOI « Mu » (synonyme de B.1.621) ;
- [40] :
- • dans la liste des VUM, ajout de C.1.2 ;
- [41] :
- • les variants Êta, Iota, Kappa ont été requalifiés de VOI à VUM le ;
[42],[43],[44],[45],[46],[47],[48],[49],[50],[51],[52],[53],[54],[55],[56],[57],[58],[59],[60],[61],[62],[63],[64],[65],[66],[67],[68],[69],[70],[71],[72],[73]
Variants préoccupants (VOC)
Les variants Alpha, Bêta, Gamma, Delta et Omicron sont classés par l'OMS comme préoccupants.
| Nom officiel de l'OMS[31] | Nextstrain | Lignées Pango[20] | GISAID[31] | Public Health England (PHE) | Date et lieu de première détection | Mutations clés[VOC 1] | Mutations en cours d’acquisition ? | |
|---|---|---|---|---|---|---|---|---|
| Alpha VOC : 2020-12-18 + au 2021-07-01[33], surveillance de S:484K et S:452R | 20I/501Y.V1 | B.1.1.7 | GRY (précédemment[31] GR/501Y.V1 | VOC-20DEC-01 précédemment[74]: VOC-202012/01[VOC 2] (first VOC of december 2020) | Royaume-Uni | ORF1a : T1001I • A1708D • I2230T • Δ3675S/3677F ; ORF1b : P314L ; S : Δ69/70 • Δ144Y • N501Y • A570D • D614G • P681H • H655Y • T716I • S982A • D1118H ; ORF8 : Q27* • R52I • Y73C ; N : D3L • R203K • G204R • S235F  Cartographie des mutations de codons du génome de SARS-CoV-2[75]. | S : S477R[Note 2]? • E484K • F490S[Note 3] • D614-(LYQDVNC)[Note 4] | |
| Bêta VOC : 2020-12-18 + au 2021-07-06[34], surveillance de S:L18F | 20H/501Y.V2[VOC 2] | B.1.351 + au 2021-07-01[33] B.1.351.2 B.1.351.3 | GH/501Y.V2 | VOC-20DEC-02 précédemment : VOC-202012/02 (second VOC of december 2020) | Afrique du Sud | ORF1a : T265I • K1655N • K3353R • Δ3675S/3677F ; ORF1b : P314L ; S : L18F • D80A • D215G • Δ241/243 • K417N • E484K • N501Y • D614G • A701V ; ORF3a : Q57H • S171L ; E : P71L ; N : T205I  Cartographie des mutations de codons du génome de SARS-CoV-2[76]. | S : V483F[Note 5]? | |
| Gamma VOC : 2021-01-11 + au 2021-07-01[33], surveillance de S:681H | 20J/501Y.V3 | P.1[VOC 2] (alias B.1.1.28.1) + au 2021-07-01[33] P.1.1 P.1.2 | GR/501Y.V3 | VOC-21JAN-02 précédemment : VOC-202101/02 | Brésil / Japon | ORF1a : S1188L • K1795Q • Δ3675S/3677F ; ORF1b : P314L • E1264D ; S : L18F • T20N • P26S • D138Y • R190S • K417N/T • E484K • N501Y • D614G • H655Y • T1027I • V1176F ; ORF3a : S253P ; ORF8 : E92K ; N : P80R • R203K • G204R ; ORF9b : Q77E  Cartographie des mutations de codons du génome de SARS-CoV-2[77]. | S : T470N[Note 6]? | |
| Delta VOI : 2021-04-04 VOC : 2021-05-11 + au 2021-07-06[34], surveillance de S:417N | 21A ou 21A/S:478K | B.1.617.2 + au 2021-07-01[33] AY.1 AY.2 | G/478K.V1 | VOC-21APR-02 | Inde | ORF1a : T3255I ; ORF1b : P314L • G662S • P1000L ; S : T19R • G142D • Δ156/157 • R158G • L452R • T478K • D614G • P681R • D950N ; ORF3a : S26L ; M : I82T ; ORF7a : V82A • T120I ; ORF8 : Δ119D/120F ; N : D63G • R203M • D377Y ; ORF9b : T60A[78] | S : A475S[Note 7]? • S477I[Note 8] ? • P479L[Note 9] ? • P479S[Note 10] ? • D614-(LYQDVNC)[Note 11] | |
| Omicron dates à préciser | lignée d'origine B.1.1.529 (alias BA.1) | 21M[80],[81] | B.1.1.529 + au 2021-12-07[53] BA.1 BA.2 | GR/484A | Afrique du Sud |
| ||
| (sous-lignée BA.1) | 21K[80] | BA.1 | GR/484A | _variant.svg) {{{1}}} | ||||
| (sous-lignée BA.2) | 21L[81] (22C pour BA.2.12.1)[83] | BA.2 | GR/484A | _variant.svg) {{{1}}} | ||||
| (sous-lignée BA.3) | — aucun clade défini —[84] | BA.3 | GR/484A | _variant.svg) Cartographie des mutations de codons du génome de SARS-CoV-2[85]. | ||||
| (sous-lignée BA.4) | 22A[86] | BA.4 | GR/484A | _variant.svg) Cartographie des mutations de codons du génome de SARS-CoV-2[85]. | ||||
| (sous-lignée BA.5) | 22B[87] | BA.5 | GR/484A | _variant.svg) Cartographie des mutations de codons du génome de SARS-CoV-2[85]. | ||||

Sources : OMS[88], PANGOlin[21], Public Health England[89], Santé Publique France[90].
Notes VOC du tableau :
Variants d'intérêt (VOI)
| Nom officiel de l'OMS[32] | Nextstrain | Lignées Pango[20] | GISAID[32] | Public Health England (PHE) | Date et lieu de première détection | Mutations clés[VOI 1] | Mutations en cours d’acquisition ? |
|---|---|---|---|---|---|---|---|
| qualification OMS : ex- VOI : 2021-03-05 requalifié VUM le 2021-09-20[41] | |||||||
| qualification OMS : ex- VOI : 2021-03-17 requalifié VUM le 2021-07-06[34] exclu de la liste des VUM le 2021-08-13[37], n'est alors plus suivi | |||||||
| qualification OMS : ex- VOI : 2021-03-17 requalifié VUM le 2021-09-20[41] | |||||||
| qualification OMS : ex- VOI : 2021-03-24 requalifié VUM le 2021-07-06[34] exclu de la liste des VUM le 2021-08-13[37], n'est alors plus suivi | |||||||
| qualification OMS : ex- VOI : 2021-03-24 requalifié VUM le 2021-09-20[41] | |||||||
| qualification OMS : ex- VOI : 2021-04-04 requalifié VUM le 2021-09-20[41] | |||||||
| Lambda VOI : 2021-06-14 | C.37 | GR/452Q.V1 | VUI-21JUN-01 | Pérou | ORF1a : T1246I • P2287S • F2387V • L3201P • T3255I • G3278S • Δ3675S/3677F • ORF1b : P314L • S : G75V • T76I • Δ246R/252G • L452Q • F490S • D614G • T859N • N : P13L • R203K G204R • G214C  Cartographie des mutations de codons du génome de SARS-CoV-2[91]. | ||
| Mu VOI : 2021-08-30 | 21H | B.1.621 | Colombie | ORF1a : T1055A • T1538I • T3255I • Q3729R • ORF1b : P314L • P1342S • S : T95I • Y144S • Y145N • R346K • E484K • N501Y • D614G • P681H • D950N • ORF3a : Q57H • ORF8 : T11K • P38S • S67F • N : T205I  Cartographie des mutations de codons du génome de SARS-CoV-2[92]. | |||
Sources : OMS[88], PANGOlin[21], Public Health England[89], Santé Publique France[90].
Notes VOI du tableau :
Variants en cours d’évaluation (VUM)
| Nom officiel de l'OMS | Nextstrain | Lignées Pango[20] | GISAID | Public Health England (PHE) | Date et lieu de première détection | Mutations clés[VUM 1] | Mutations en cours d’acquisition ? |
|---|---|---|---|---|---|---|---|
| ex- VOI : 2021-03-05 requalifié VUM le 2021-07-06[34] | 20C/S:452R | B.1.427/B.1.429 | GH/452R.V1 | États-Unis | ORF1a : T265I • I4205V • ORF1b : P314L • D1183Y • S : S13I • W152C • L452R • D614G • ORF3a : Q57H • ORF8 : V100L • N : T205I • M234I  Cartographie des mutations de codons du génome de SARS-CoV-2[93]. | ||
| qualification OMS : ex- VOI : 2021-03-17 requalifié VUM le 2021-07-06[34] exclu de la liste des VUM le 2021-08-13[37] : n'est alors plus suivi | |||||||
| ex- VOI : 2021-03-17 | 20C | B.1.525 | G/484K.V3 | VUI-21FEB-03 | Royaume-Uni / Nigéria | ORF1a : T2007I • Δ3675S/3677F • ORF1b : P314F • S : Q52R • A67V •Δ69H/70V • Δ144Y • E484K • D614G • Q677H • F888L • E : L21F • M : I82T • ORF6 : Δ2F • N : S2M • D3Y • A12G • T205I • ORF9b : H9D  Cartographie des mutations de codons du génome de SARS-CoV-2[94]. | |
| qualification OMS : ex- VOI : 2021-03-24 requalifié VUM le 2021-07-06[34] exclu de la liste des VUM le 2021-08-13[37] : n'est alors plus suivi | |||||||
| ex- VOI : 2021-03-24 (S477N ou E484K) | 21F | B.1.526 | GH | New York City | ORF1a : T265I • L3201P • Δ3675S/3677F • ORF1b : P314L • Q1011H • S : L5F • T95I • D253G • S477N ou E484K (au choix) • D614G • ORF3a : P42L • Q57H • ORF8 : T11I Cartographie des mutations de codons du génome de SARS-CoV-2[95]. | ||
| ex- VOI : 2021-04-04 | 21B | B.1.617.1 | G/452R.V3 | VUI-21APR-01 | Inde | ORF1a : T1567I • T3646A • ORF1b : P314L • G1129C • M1352I • K2310R • S2312A • S : L452R • E484Q • D614G • P681R • Q1071H • ORF3a : S26L • M : I82S • ORF7a : V82A • N : R203M • D377Y  Cartographie des mutations de codons du génome de SARS-CoV-2[96]. | |
| (20A) | B.1.620 | Afrique | ORF1a : T403I • V1991I • Δ3675S/3677F • ORF1b : P314L • A1215S • S : P26S • Δ69H/70V • V126A • Δ144Y • Δ241/243 • H245Y • S477N • E484K • D614G • P681H • T1027I • D1118H • ORF7b : Δ144L • N : A220V • ORF9b : I5T | ||||
| (20D) | C.36.3 | Asie | ORF1a : E102K • T1246I • D1639N • D2980N • D3222N • G3278S • S3687L • L3691S • T4090I • ORF1b : P314L • D1028Y • S : S12F • W152R • D614G • Q677H • M : I82T • ORF7b : A43S • N : R203K • G204R • G212V | S : Q474H[Note 12] ? | |||
| (20B) | B.1.1.318 | Afrique | ORF1a : K2511N • T2936I • A3209V • Δ3675S/3677F • ORF1b : P314L • V2371M • S : T95I • Δ144Y • E484K • D614G • P681H • D796H • M : I82T • ORF7b : *44X • ORF8 : Δ1M/2K • F3X • E106X • *122X • N : R203K • G204R • Δ208A • R209G | ||||
| (20B) | AT.1 | Russie | ORF1a : L39K • S376L • V1006F • T1093A • T2247N • T3255I • Q3729R • S4119T • ORF1b : A190S • P314L • T1173N • V1905L • A2431V • S : P9L • Δ136C/144Y • D215G • H245P • E484K • D614G • E780K • ORF3a : L95M • M : L16I • N : R203K • G204R | ||||
| (20B) | B.1.1.519 | Mexique | ORF1a : P959S • I3618V • T4175I • ORF1b : P314L • S : T478K • D614G • P681H • T732A • N : R203K • G204R | S : D614-(LYQDVNC)[Note 13] | |||
Sources : OMS[88], PANGOlin[21], Public Health England[89], Santé Publique France[90].
Notes VUM du tableau :
Variants non-suivis
Ce sont les variants, précédemment classifiés comme VOI, qui ont été successivement requalifiés de VOI à VUM, puis de VUM à non-suivi (noté ici NS)
| Nom officiel de l'OMS | Nextstrain | Lignées Pango[20] | GISAID | Public Health England (PHE) | Date et lieu de première détection | Mutations clés[NS 1] | Mutations en cours d’acquisition ? |
|---|---|---|---|---|---|---|---|
| ex- VOI : 2021-03-17 requalifié VUM le 2021-07-06[34] exclu de la liste des VUM le 2021-08-13[37], n'est alors plus suivi | 20J | P.2 (alias B.1.1.28.2) | GR | VUI-21JAN-01 | Brésil | ORF1a : L3468V • L3930F • ORF1b : P314L • S : E484K • D614G • V1176F • N : A119S • R203K • G204R • M234I  Cartographie des mutations de codons du génome de SARS-CoV-2[97]. | |
| ex- VOI : 2021-03-24 requalifié VUM le 2021-07-06[34] exclu de la liste des VUM le 2021-08-13[37], n'est alors plus suivi | 21E | P.3 | GR | VUI-21MAR-02 | Philippines | ORF1a : D1554G • L3201P • D3681E • L3930F • ORF1b : P314L • A1291V • S : Δ141L/143V • E484K • N501Y • D614G • P681H • E1092K • H1101Y • V1176F • ORF8 : K2Q • N : R203K • G204R  Cartographie des mutations de codons du génome de SARS-CoV-2[98]. |
Sources : OMS[88], PANGOlin[21], Public Health England[89], Santé Publique France[90].
Notes NS du tableau :
_(cropped).jpg)


